A novel framework for evaluating the performance of codon usage bias metrics
Abstract
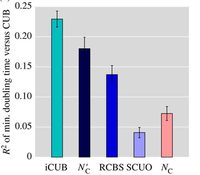
The unequal utilization of synonymous codons affects numerous cellular processes including translation rates, protein folding and mRNA degradation. In order to understand the biological impact of variable codon usage bias (CUB) between genes and genomes, it is crucial to be able to accurately measure CUB for a given sequence. A large number of metrics have been developed for this purpose, but there is currently no way of systematically testing the accuracy of individual metrics or knowing whether metrics provide consistent results. This lack of standardization can result in false-positive and false-negative findings if underpowered or inaccurate metrics are applied as tools for discovery. Here, we show that the choice of CUB metric impacts both the significance and measured effect sizes in numerous empirical datasets, raising questions about the generality of findings in published research. To bring about standardization, we developed a novel method to create synthetic protein-coding DNA sequences according to different models of codon usage. We use these benchmark sequences to identify the most accurate and robust metrics with regard to sequence length, GC content and amino acid heterogeneity. Finally, we show how our benchmark can aid the development of new metrics by providing feedback on its performance compared to the state of the art.