NullSeq: A tool for generating random coding sequences with desired amino acid and GC contents
Abstract
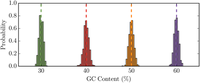
The existence of over- and under-represented sequence motifs in genomes provides evidence of selective evolutionary pressures on biological mechanisms such as transcription, translation, ligand-substrate binding, and host immunity. In order to accurately identify motifs and other genome-scale patterns of interest, it is essential to be able to generate accurate null models that are appropriate for the sequences under study. While many tools have been developed to create random nucleotide sequences, protein coding sequences are subject to a unique set of constraints that complicates the process of generating appropriate null models. There are currently no tools available that allow users to create random coding sequences with specified amino acid composition and GC content for the purpose of hypothesis testing. Using the principle of maximum entropy, we developed a method that generates unbiased random sequences with pre-specified amino acid and GC content, which we have developed into a python package. Our method is the simplest way to obtain maximally unbiased random sequences that are subject to GC usage and primary amino acid sequence constraints. Furthermore, this approach can easily be expanded to create unbiased random sequences that incorporate more complicated constraints such as individual nucleotide usage or even di-nucleotide frequencies. The ability to generate correctly specified null models will allow researchers to accurately identify sequence motifs which will lead to a better understanding of biological processes as well as more effective engineering of biological systems.